
Top 5 Most Revolutionary Uses of Single-Cell RNA Sequencing
The use of single-cell RNA sequencing (scRNA-seq) in biomedical research has skyrocketed in recent years. The reason for the growing popularity of this technology is that scRNA-seq enables researchers to understand the cellular heterogeneity and functional diversity in a sample by analyzing transcriptome profiles of individual cells.
This has led to new and ground-breaking discoveries in several fields, including development, oncology, immunology, neuroscience, and the study of rare diseases. In this blog post, we take a detailed look at some of these recent breakthroughs through the lens of scRNA-seq applications (outlined in Figure 1)!
Read more about the development and rise of scRNA-seq at https://www.biomage.net/blog/riseofscrnaseq

Figure 1. Applications of scRNA-seq. The figure outlines the most common applications of scRNA-seq in biological research.
1. Cellular heterogeneity
One of the major strengths of single-cell analysis is that heterogeneity within and between samples can easily be investigated. It means that you can detect differences between different cell populations and between the same cell population across different samples. Cellular heterogeneity can encompass genotypic or phenotypic differences due to genetic variation, a mixture of different cell types, differentiation and more. This is often used to broaden our knowledge about the role of heterogeneity in disease.
For example, scRNA-seq has been used to uncover the heterogeneity of T cells in cancer, with the goal to investigate the host’s immune response to a tumor. Guo et al. analyzed thousands of T cells derived from non-small lung cancer [1]. They discovered the presence of different T cell subsets – pre-exhausted and exhausted cytotoxic T cells, inter-tissue effector T cells, and T regulatory cells. Interestingly, T regulatory cells displayed further heterogeneity and were characterized by a bimodal distribution of TNFRSF9 (an activation marker). This hints at the existence of recently activated and resting T regulatory cells. These findings are important for the efficacy of cancer immunotherapies and the prognosis, as different T cell compositions in the tumor relate to different clinical behaviors.
Our ultimate understanding of cellular heterogeneity in entire tissues or organs is captured in cell atlases. This application of scRNA-seq is now the backbone of a global scientific collaboration to create a human cell atlas [2]. The aim of this atlas is to map the position, function and characteristics of each healthy cell in the human body on a transcriptional level. The human cell atlas also strives to represent various ages, genders and lifestyles to illustrate the diversity in human biology. When complete it would provide reference maps for further research.
2. Rare cell types
By combining high throughput analysis with a highly sensitive assessment of gene expression profiles, scRNA-seq is the best practice for analyzing rare and unknown cell populations. Identifying different cell types in a population can lead to a better understanding of the function of cells or tissues. It can also pinpoint novel interactions between cells or potential pathological mechanisms.
In a ground-breaking study, using scRNA-seq researchers identified a population of rare cells in the lungs, called pulmonary ionocytes, that are thought to be responsible for causing cystic fibrosis (Figure 2). These findings reframed our understanding of airway diseases such as asthma and cystic fibrosis. The single-cell atlas identified a new rare cell type – Foxil1+ pulmonary ionocytes [3]. Knockout of Foxil1 in mouse ionocytes caused a loss of cystic fibrosis transmembrane conductance regulator (Cftr) expression and led to disrupted airway fluid and mucus physiology – a common characteristic of cystic fibrosis. This allowed the creation of a new cellular narrative for airway disease by relating expression programs specific to cell type to key disease genes.

Figure 2. A single-cell expression atlas of mouse tracheal epithelial cells. a) Overview of the experiment and analysis. b) T-distributed stochastic neighbor embedding (t-SNE) coloured by cell type. The novel ionocyte cluster is circled. Montoro et al., 2018.
3. Developmental biology and differentiation
scRNA-seq has also proved revolutionary in the field of developmental biology by providing deep insight into cells as they differentiate. It’s not just about broadening our knowledge of who we are, but it also gives insights into congenital disorders and regenerative medicine. During development, each cell has its own distinct cell fate after differentiation, and scRNAseq gives new insights into these differential states and trajectories during development.
You can look at the cell’s individual differentiation pathways and the cell type characteristic differentiation, and the underlying mechanisms behind those two. During development, cells acquire distinct fates by transitioning through different transcriptional states. So with scRNA-seq, you can “take snapshots” of a cell during differentiation, which then can be analyzed using trajectory inference algorithms. This allows to place cells following the same differentiation process along a pseudotime trajectory. Pseudotime analysis often results in tree-like models with potential cell fate decision points as cells often go through different decision moments before differentiating towards a particular lineage. Figure 3 shows an example of cell fate analysis by Farrell et al. of early zebrafish embryogenesis [4]!
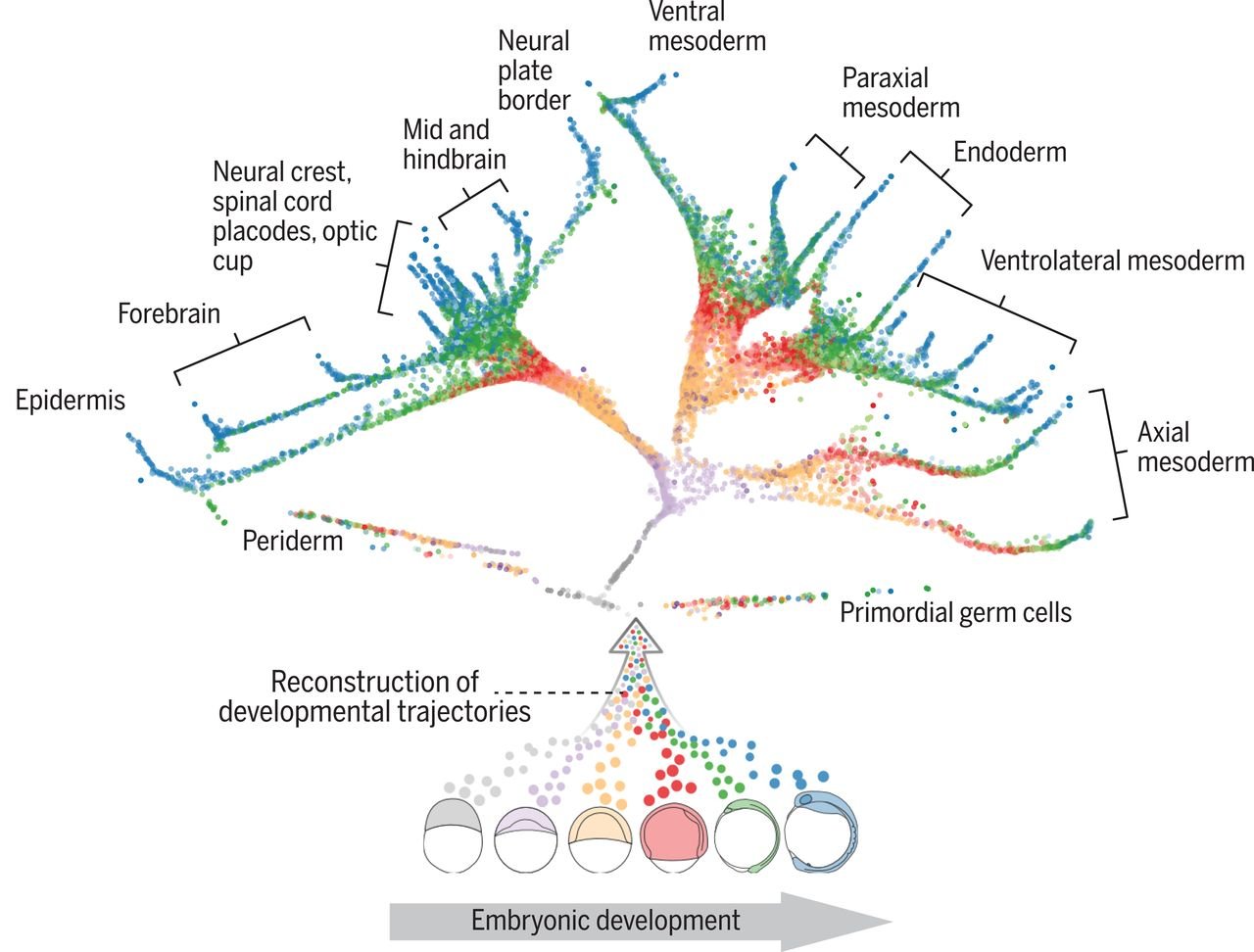
Figure 3. The developmental tree of early zebrafish embryogenesis. Transcriptomes were obtained from zebrafish embryos at 12 different developmental stages. The trajectories show a reconstruction of cell fates of 25 cell types. Each individual cell is represented by a point and coloured by the developmental stage. Farrell et al., 2018.
Farrell et al. investigated 12 stages of early zebrafish embryogenesis at a single-cell resolution [4]. Importantly, they discovered that not all cell fate decisions might be binary - a cell could have more than two possible fates. For example, the first molecular specification may separate not only the germ layers but also the axial and nonaxial mesendoderm. They also found cells in intermediate developmental states which expressed genes characteristic of multiple downstream cell fates. This suggests the idea of plasticity between developmental states.
Using scRNAseq in developmental biology provides insight into the developmental errors behind congenital diseases. It’s possible to study the embryos in developmental mutant models at a single-cell resolution. This gives detail on the faulty mechanisms underlying developmental disorders.
Congenital heart defects are the most common type of birth anomalies, and scRNA-seq allowed us to discover the exact faulty mechanisms in individual cells in this condition. Transcriptional changes in early cardiac progenitor cells were investigated during normal and abnormal cardiogenesis [5]. The disruption of cardiac progenitor cells results in congenital heart defects. Lineage-specifying transcription factor Hand2 determined outflow tract cells. And the loss of Hand2 led to improper differentiation of cells and, thus, a disrupted cardiac development. This study is a crucial step toward developing preventative approaches for congenital heart defects and postnatal interventions.
4. Drug targets
Its high sensitivity and resolution make scRNA-seq an effective way to identify molecules and pathways that are associated with disease and, consequently, have the potential to become biomarkers or drug targets.
For example, the discovery of new biomarkers is crucial to the detection of early-stage cancers, as early diagnosis improves the survival rate. In a recent study, scRNA-seq was used to identify new candidate genes for the diagnosis of early-stage lung cancer [6]. It was found that the differential expression of microRNAs that regulate chemokine (like CXCL1) mRNA expressions in lung adenocarcinoma patient samples can be used as a molecular biomarker.
Biomarkers can also be potential targets for new treatments and thus present great pharmaceutical interest. In more complex diseases, therapeutics usually consist of multiple drug targets, therefore identification of new markers and targets is of uttermost importance.
Wang et al. showed a previously underappreciated role of epidermal growth factor receptor mutant (EGFRvIII) and its associated oncogene in glioblastoma [7]. EGFRvIII mutant cells lack the extracellular ligand-binding domain, and this alteration accounts for just about 30% of glioblastoma cases. RAD51AP1 oncogene was highly upregulated in EGFRvIII-positive cells. Knocking down RAD51AP1 led to a reduced tumor volume and prolonged survival time in the EGFRvIII glioblastoma model. So, it is thought that combining temozolomide (the main chemotherapeutic drug used to treat glioblastoma) and RAD51AP1 could enhance the therapeutic effect of the treatment and prolong patient survival.
5. Effects of existing drugs
Candidate drugs can lead to changes in gene expression, and this can be studied with scRNA-seq. So, it allows us to look at how a particular treatment affects each cell type separately. scRNA-seq also allows screening for the effects of drugs already in use. This can lead to the discovery of new purposes for existing treatments, as well as the identification of dangerous off-target effects that otherwise could have been missed in preclinical studies.
In line with this, single-cell technology has enabled researchers to discover the molecular pathways behind treatment resistance in cancer. scRNAseq also allows the identification and characterization of individual cells or cell groups that have become resistant to targeted drugs. This can lead to the design of better therapeutic strategies for cancer patients.
For instance, scRNA-seq has been used in metastatic human breast cancer cells treated with paclitaxel – a chemotherapeutic agent [8]. The study revealed a small number of cells that failed to respond to the drug by producing specific RNA variants in genes involved in microtubule organization and stabilization, cell adhesion and cell surface signaling. These variants were otherwise undetected in cells not treated with chemotherapy. This shows the reversible cellular stress response that drives the heterogeneity of gene expression within a population by cell-specific RNA variants.
Conclusions
To conclude, the extent of research fields applying scRNA-seq is very broad and is continuing to widen. Scientists have taken advantage of scRNA-seq to solve long-standing problems. As each cell in an organ or a tissue plays a role in the overall function of the organism, single-cell sequencing allows us to look at this specific contribution of each cell. This has revealed new insights into disease pathology and novel targets for treatment development. Such insights are transforming our understanding of pathogenesis and the involvement of genetic components in conditions such as cystic fibrosis and asthma.
We at Biomage look forward to more remarkable discoveries coming out of single-cell studies!
References:
[1] Guo, X., Zhang, Y., Zheng, L., Zheng, C., Song, J., Zhang, Q., Kang, B., Liu, Z., Jin, L., Xing, R. and Gao, R., 2018. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nature medicine, 24(7), pp.978-985.
[2] https://www.humancellatlas.org
[3] Montoro, D.T., Haber, A.L., Biton, M., Vinarsky, V., Lin, B., Birket, S.E., Yuan, F., Chen, S., Leung, H.M., Villoria, J. and Rogel, N., 2018. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature, 560(7718), pp.319-324.
[4] Farrell, J.A., Wang, Y., Riesenfeld, S.J., Shekhar, K., Regev, A. and Schier, A.F., 2018. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science, 360(6392), p.eaar3131.
[5] de Soysa, T.Y., Ranade, S.S., Okawa, S., Ravichandran, S., Huang, Y., Salunga, H.T., Schricker, A., Del Sol, A., Gifford, C.A. and Srivastava, D., 2019. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature, 572(7767), pp.120-124.
[6] Kim, J., Xu, Z. and Marignani, P.A., 2021. Single-cell RNA sequencing for the identification of early-stage lung cancer biomarkers from circulating blood. NPJ genomic medicine, 6(1), pp.1-15.
[7] Wang, Q., Tan, Y., Fang, C., Zhou, J., Wang, Y., Zhao, K., Jin, W., Wu, Y., Liu, X., Liu, X. and Kang, C., 2019. Single-cell RNA-seq reveals RAD51AP1 as a potent mediator of EGFRvIII in human glioblastomas. Aging (Albany NY), 11(18), p.7707.
[8] Lee, M.C.W., Lopez-Diaz, F.J., Khan, S.Y., Tariq, M.A., Dayn, Y., Vaske, C.J., Radenbaugh, A.J., Kim, H.J., Emerson, B.M. and Pourmand, N., 2014. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proceedings of the National Academy of Sciences, 111(44), pp.E4726-E4735.